Exploring the Structural Biology of Cancer
Cells in our bodies have evolved to work together, constantly communicating with each other, sharing resources, and growing and dividing only when and where needed. Normal cells that become transformed into cancer cells go rogue and grow without this cooperation with other cells in the body. "Cancer" encompasses many different diseases, but cancer cells share similar characteristics, including ignoring normal controls on growth, evading the body's built-in defenses, tuning metabolism and their surrounding environment for faster growth, and in the most severe cases, invading other parts of the body. Cells typically gain these characteristics through changes in their genomes.
Structural biologists are studying all aspects of this process, including the mechanisms that underpin damage to and repair of the genome, the way that these genetic changes lead to cancer, and ways for us to use this knowledge to discover and develop new treatments to fight cancer.
Access the individual sections in this resource
- Oncogenes encode proteins that are involved in malignant transformation
- Carcinogens damage the genome
- Cancer cells ignore the normal signals that control growth
- Cancer cells modify themselves and their environment to support growth
- Cancer cells evade the normal protections of the cell
- Cancers spread by gaining the ability to invade other parts of the body
- Cancer chemotherapy often targets cells that grow quickly
- Targeted therapies attack cancer cells more precisely
- Additional Resources
1. Oncogenes encode proteins that are involved in malignant transformation
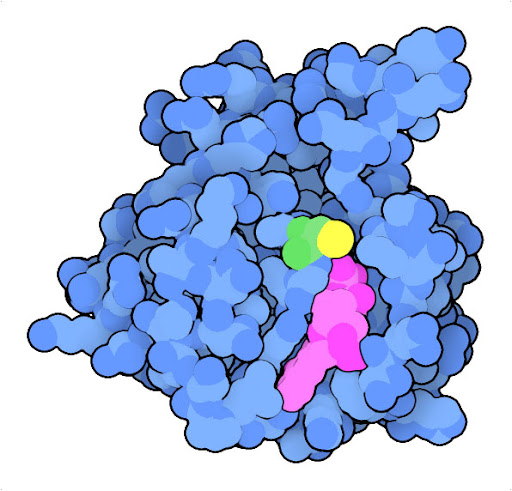
Mutation of glycine to cysteine at position 12 (green, with sulfur in yellow) in Ras protein leads to a protein that is continually activated. The structure of the oncogenic mutant (PDB ID 4ldj) reveals that the mutation modifies the interaction with GDP (magenta) and GTP, which act as the chemical switch that turns the protein on and off.
Oncogenes are key players in malignant transformation of normal cells into cancer cells. A gene can become an oncogene if it changes the properties of the cell in a way that leads to uncontrolled growth. Many types of normal cellular genes can become oncogenes, expressing oncoproteins that may be signaling proteins that regulate growth, proteins that protect us from cancer cells that have undergone genomic alteration, and variant proteins that no longer work correctly or are made at unnatural levels.
The Ras protein, for example, is a central player in signaling processes that normally regulate cell growth. In normal cells, Ras relays signals from receptors embedded in the outer membrane to many intracellular proteins contributing to cellular structure and growth. In many cancer cells, the Ras oncogene encodes a variant of the protein, such as the G12C variant shown here, that is continually active, promoting growth without the need for activation by a receptor.
The G12C mutated form of the Ras oncogene codes for an overly-active protein causing uncontrolled proliferation, but our cells possess other proteins that can stop these oncogenes in their tracks. The best known of these tumor suppressor proteins, p53, is a transcriptional regulator that normally senses DNA damage and initiates DNA repair and processes like apoptosis to clear up the problem. However, the gene for p53 tumor suppressor is commonly mutated in cancer cells, encoding an inactive form and blocking this mode of self protection.
2. Carcinogens damage the genome
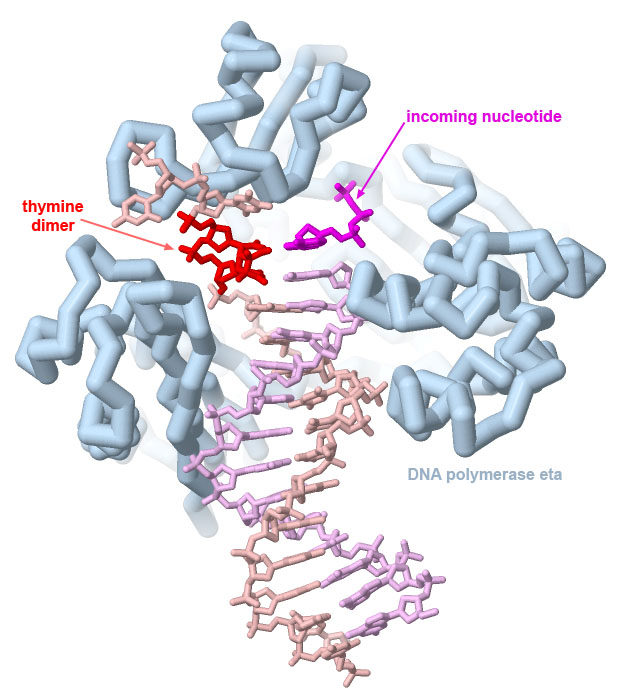
DNA polymerase eta can correctly replicate DNA strands with UV-induced thymine dimers, as seen in PDB ID 1rys. Dimers formed with cytosine bases, however, are often not copied correctly, leading to characteristic C to T mutations caused by UV damage.
The structure reveals that it is able to copy through damaged sections of the DNA because the overall complex is smaller and looser than the major polymerases involved in DNA replication (and consequently, not as accurate). Image created with Jmol.
Genes are often transformed into oncogenic forms through damage by carcinogens. Many carcinogens cause localized chemical changes in the genome. For example, ultraviolet light causes neighboring thymine (or cytosine) nucleotides to react chemically with one another. The resultant nucleotide dimer is difficult for the DNA replication machinery to copy correctly and leads to characteristic C to T mutations that are observed in UV-induced DNA damage. Reactive compounds, such as many components of tobacco smoke, also damage DNA bases and can lead to mutations when the DNA is replicated or transcribed into mRNA.
Because damage to the genome is so dangerous, cells have various specialized molecules and macromolecular assemblies that repair problems with the DNA. These molecular machines include single proteins that repair individual damaged nucleotides, such as photolyases. For more substantial DNA damage, drastic mechanisms are employed: nucleotide excision repair cuts out a piece of the genome and then backfills it with a new strand, and non-homologous end joining reconnects double-strand breaks in the DNA. When the damage becomes too much for cells to correct, they initiate the process of apoptosis or programmed death and kill themselves.
3. Cancer cells ignore the normal signals that control growth
Cells constantly monitor their surroundings, deciding together when they need to grow and divide for the good of the whole body. Three types of proteins control this communication. First, messages are passed from cell to cell using small proteins, such as growth factors and cytokines. These proteins are secreted from one cell, then diffuse to neighboring cells. There, they are recognized by specific receptor proteins that have the job of “transducing” the message from the outside of the cell to the cytoplasm inside. Many of these receptors have two functionally different segments with different structures: a growth-factor-binding domain outside the cell, and a kinase catalytic domain inside the cell. When the right growth factor binds, it brings together two or more receptors, allowing the kinases inside to phosphorylate and thereby activate each other. Then, the active kinase domains in turn activate many other signaling proteins within the cell, such as membrane-associated ras protein shown above. A cascade of activation is initiated, which produces many active kinases that activate other proteins throughout the cell. The signal from the growth factor eventually reaches the nucleus, where it changes the genetic program of the cell by increasing expression of mRNA encoding proteins required for growth and down-regulating expression of other proteins that are normally present to control growth. Growth factor signaling can also have direct effects on proteins in the cytoplasm that in turn regulate protein synthesis by ribosomes.
When these growth signals become corrupted, cells may gain unwanted permission to divide and multiply, proliferating to form a primary tumor. Many oncogenes are part of the signaling processes that mediate this type of communication, such as the MAPK (or mitogen-activated protein kinase) signaling pathway shown here. In some cases, the proteins mutate so that they are always activated, continually sending the signal to grow. In other cases, the mutation can inactivate a protein that normally slows growth, taking off the brakes.
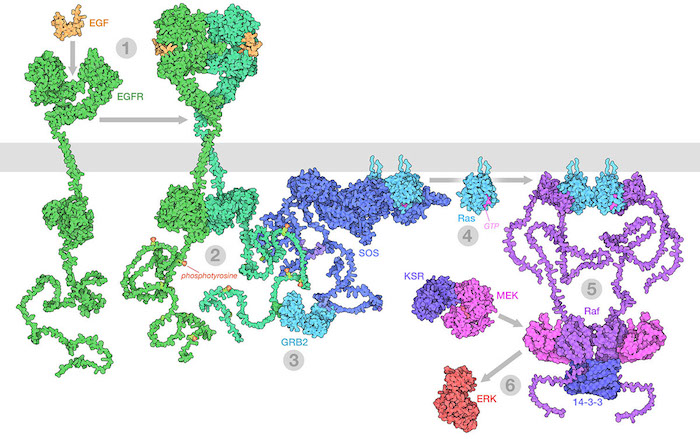
The Raf/MEK/ERK (MAPK) pathway is one of the ways that growth signals are disseminated inside the cell. 1) Epidermal growth factor (EGF) binds to its receptor (EGFR), causing it to dimerize. 2) The catalytic domains of EGFR phosphorylate tyrosines on the long unstructured EGFR tails. 3) GRB2 recognizes the phosphorylated tyrosines and recruits SOS. 4) SOS replaces GDP with GTP in Ras proteins. 5) The activated Ras forms nanoclusters that activate Raf, with the help of 14-3-3 protein. 6) Raf phosphorylates MEK, which is normally held in an inactive complex with KSR. Active MEK then phosphorylates ERK, a kinase that will activate many processes throughout the cell. Illustration created from decades of structural studies in PDB IDS 1egf, 1nql, 1m17, 2jwa, 3njp, 2gs6, 1gri, 1xd2, 3ksy, 5p21, 6xi7, 6q0j, 2y4i, 1pme, and unstructured chains from AlphaFold2.
4. Cancer cells modify themselves and their environment to support growth
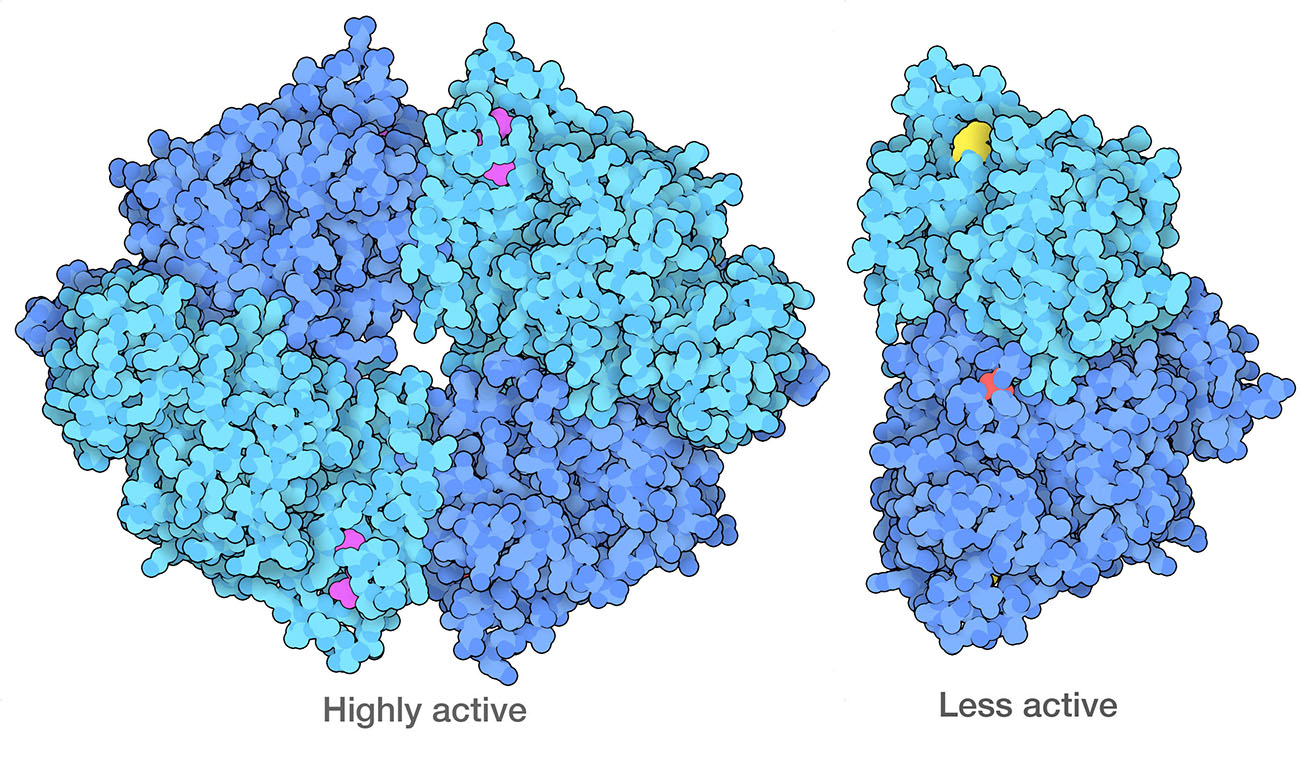
Cancer cells have modified forms of PKM2 (pyruvate kinase 2) that form dimers, which are less active than the normal tetrameric form. One of the ways they do this is to modify specific amino acids, including a serine (red) that is phosphorylated and a lysine (yellow) that is acetylated. PDB ID 4fxf and 6wp3.
Normal physiological processes do not support unnatural growth of malignant tumors, so cancer cells often make changes to benefit their growth. For example, as a tumor grows, the interior cells become progressively separated from the circulatory system and would be increasingly starved for oxygen and nutrients. Cancer cells often solve this problem by releasing signaling molecules, such as vascular endothelial growth factor or VegF that stimulate the growth of new blood vessels. These small proteins mobilize the body’s normal processes for building new vasculature, remodeling the circulatory system for their own benefit.
Cancer cells also reprogram their own metabolic processes to support the enormous energy needs of their uncontrolled growth. For example, cancer cells often ramp up the use of glycolysis, while suppressing aerobic processes for energy production. This phenomenon might seem like a paradox: why would a rapidly-dividing cell switch to a less efficient method for generating energy? The answer is that glycolysis is also used to create many of the starting materials for building amino acids and nucleotides, which are needed to build new proteins and nucleic acids for the rapidly dividing cell. The enzyme that performs the final step of glycolysis, pyruvate kinase, acts as a gatekeeper to help make this decision.
5. Cancer cells evade the normal protections of the cell
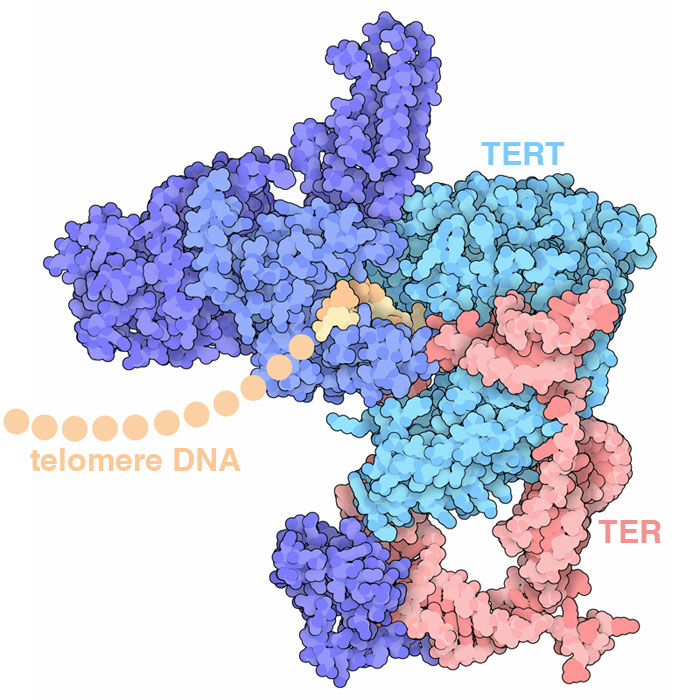
Telomerase adds DNA to the end of chromosomes, repeatedly adding a 6-base-pair sequence of nucleotides to form a “telomere”. The structure (PDB ID 6d6v) reveals how several protein chains work together with a specialized RNA to perform this task: the TER RNA acts as the template for arranging incoming nucleotides and the TERT protein performs the reaction.
Cells possess many powerful mechanisms for protecting and repairing themselves, plus a mechanism for self-destruction if the danger or damage is too great for recovery. Cancer cells often exhibit genetic changes that evade these protections. Apoptosis, also called programmed cell death, is central for orderly self-destruction of unwanted cells. When initiated by DNA damage sensors like MDM2, regulatory proteins such as the p53 tumor suppressor act to increase production of apoptotic proteins. In the initial steps, an apoptosome is built, which in turn activates caspases and nucleases that systematically dismantle the cell.
Our cells also contain a hard-wired process that limits the number of times it can divide. Every time a cell divides, each of its chromosomes becomes shortened because the DNA replication machinery cannot copy to the end of the DNA strand. To solve this problem, our chromosomes have long, repeated sequences of nucleotides at the end, called telomeres, that don't encode proteins and thus don't cause problems if they're not replicated. However, once the number of telomere repeats falls below a critical level the cell can no longer divide. Cancers need to circumvent this limitation in order to continue growing. Telomerase is a key part of this limitation. It adds new DNA to the ends of chromosomes. In normal adult cells, telomerase is not present, so cells “age out” after a defined number of DNA replications (typically 50-70). Cancer cells, on the other hand, often make telomerase, allowing them to keep replicating indefinitely.
6. Cancers spread by gaining the ability to invade other parts of the body
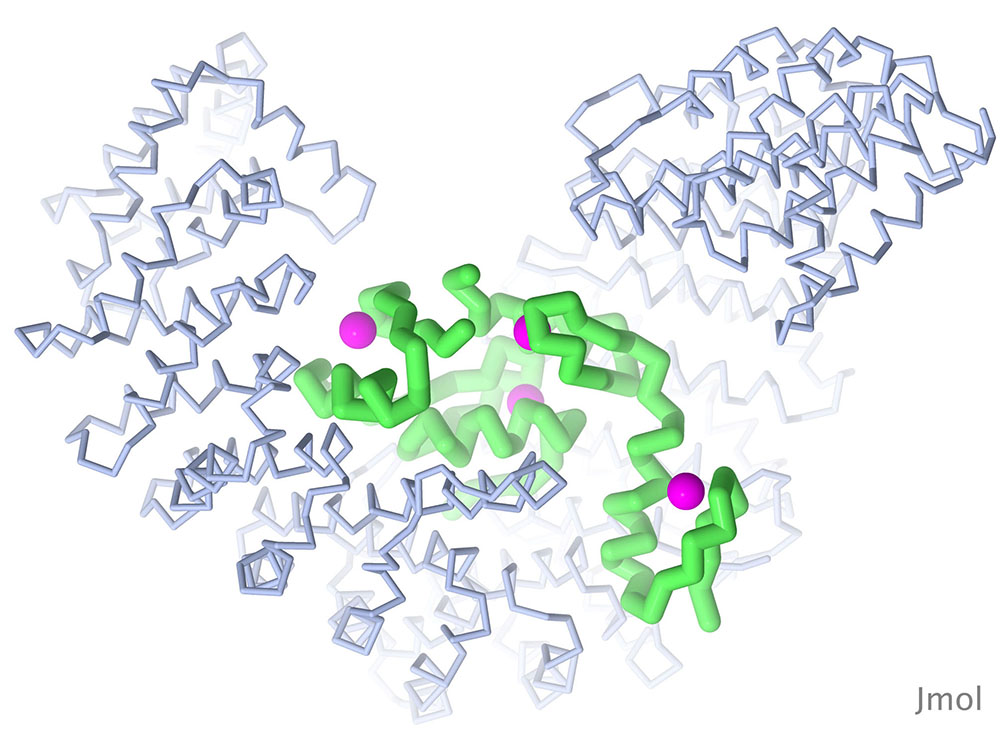
The Snail protein is one of several transcription factors that control the transition of embryonic cells from epithelial to mesenchymal types. The structure shown here (PDB ID 3w5k) includes the DNA-binding portion of the molecule, which includes four zinc fingers, bound to importin beta, which is needed to transport Snail into the nucleus. Snail is in green, zinc in magenta, and importin beta in blue. Image created with Jmol.
Most cancer deaths are not caused by the primary tumor. Instead, they occur when cancer cells migrate or metastasize. Metastatic cells gain the ability to separate from a tumor, travel through the bloodstream, and colonize other parts of the body, ultimately forming new tumors in different organs. Mechanisms underpinning metastasis remain a subject of intensive study. One key process is thought to involve a process that is normally activated during development of an embryo.
The epithelial-mesenchymal transition (EMT) allows embryonic cells to change their motility and invasiveness in ways that are needed for formation of complex structures in our tissues and organs, such as the growth of the complex shapes and connections of the nervous system. Cancer cells can also activate this process to enable their own invasive properties. The protein shown here (Snail) is a transcription factor that regulates EMT and is associated with cancer pathogenesis.
7. Cancer chemotherapy often targets cells that grow quickly
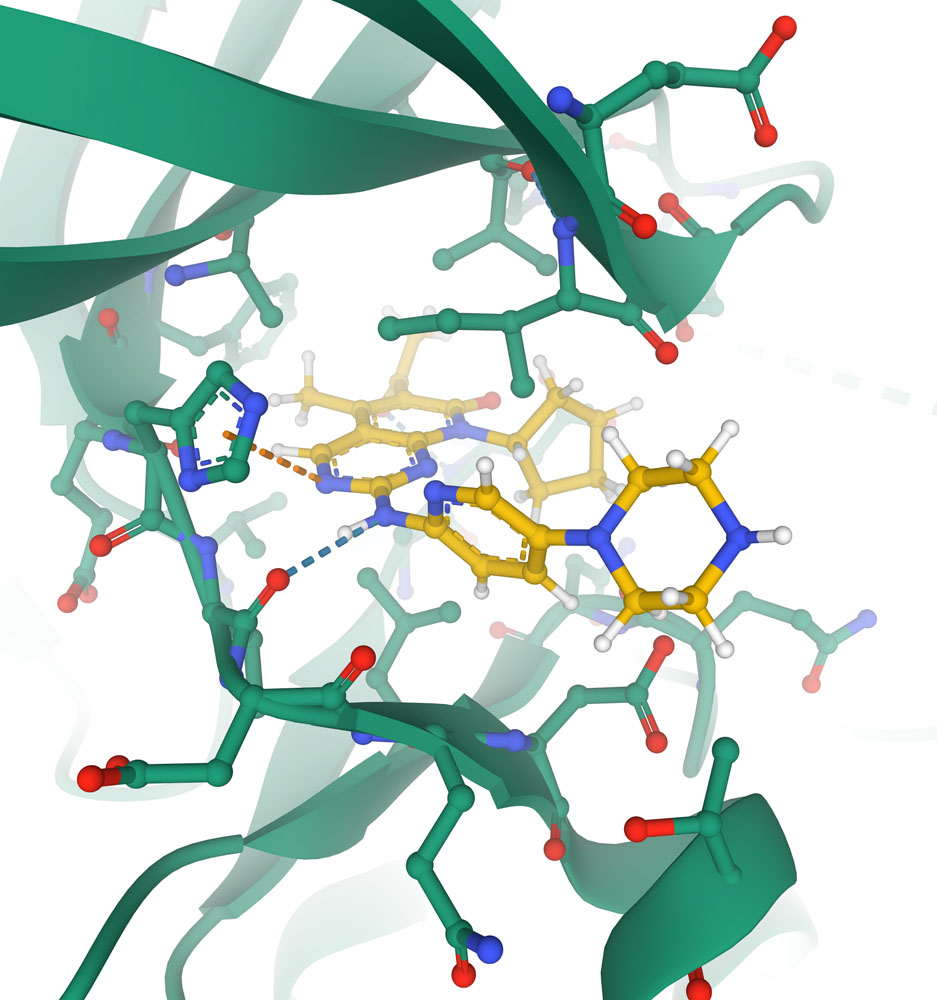
Close-up of the active site of the cyclin-dependent kinase CDK6 (shown with carbon atoms in green) with the chemotherapeutic agent palbociclib (shown with carbon atoms in yellow) blocking the active site. Key interactions with protein are shown with dotted lines in this specialized view provided in Mol* (PDB ID 5l2i).
In order to treat cancer, oncologists need ways to selectively kill cancer cells. If the tumor is localized, they take the direct approach and use surgery to remove it or focused ionizing radiation to kill it in place. Once tumor cells have spread, chemotherapy is used. The central feature of cancer cells is that they grow abnormally fast, so many chemotherapeutic drugs target cells that are rapidly dividing, either by killing the dividing cells or stopping their growth. Unfortunately, cell killing or cytotoxic chemotherapeutic drugs also kill cells that normally divide rapidly (e.g, skin cells, hair follicles, gastrointestinal epithelium), leading to unwelcome side effects like rash, hair loss and nausea.
Many different processes in cells are targeted by chemotherapeutic drugs. Cytotoxic drugs fall into a few classes. Drugs like alkylating agents and cisplatin target DNA, making chemical modifications to the DNA bases and ultimately blocking replication of the cell genome. Antimetabolite drugs block key steps in metabolism, shutting the cell down. Methotrexate, for example, blocks formation of a key cofactor needed by many enzymes. Several alkaloid molecules from plants block different stages of the cell cycle. For example, placitaxel binds to microtubules and blocks the separation of chromosomes during cell division. Antibiotics such as daunorubicin and doxorubicin bind to DNA and block the essential action of topoisomerases.
Structural biology has played a central role in the understanding of these drugs, and the discovery of new and improved ones. Some drugs were discovered from natural sources, such as paclitaxel from the bark of yew trees. Other drugs are the result of years of study, building on insights from structures of the target molecules. For example, structure-guided design has led to the discovery of new drugs to block kinases involved in regulation of growth, such as the cyclin-dependent kinase shown here.
8. Targeted therapies attack cancer cells more precisely
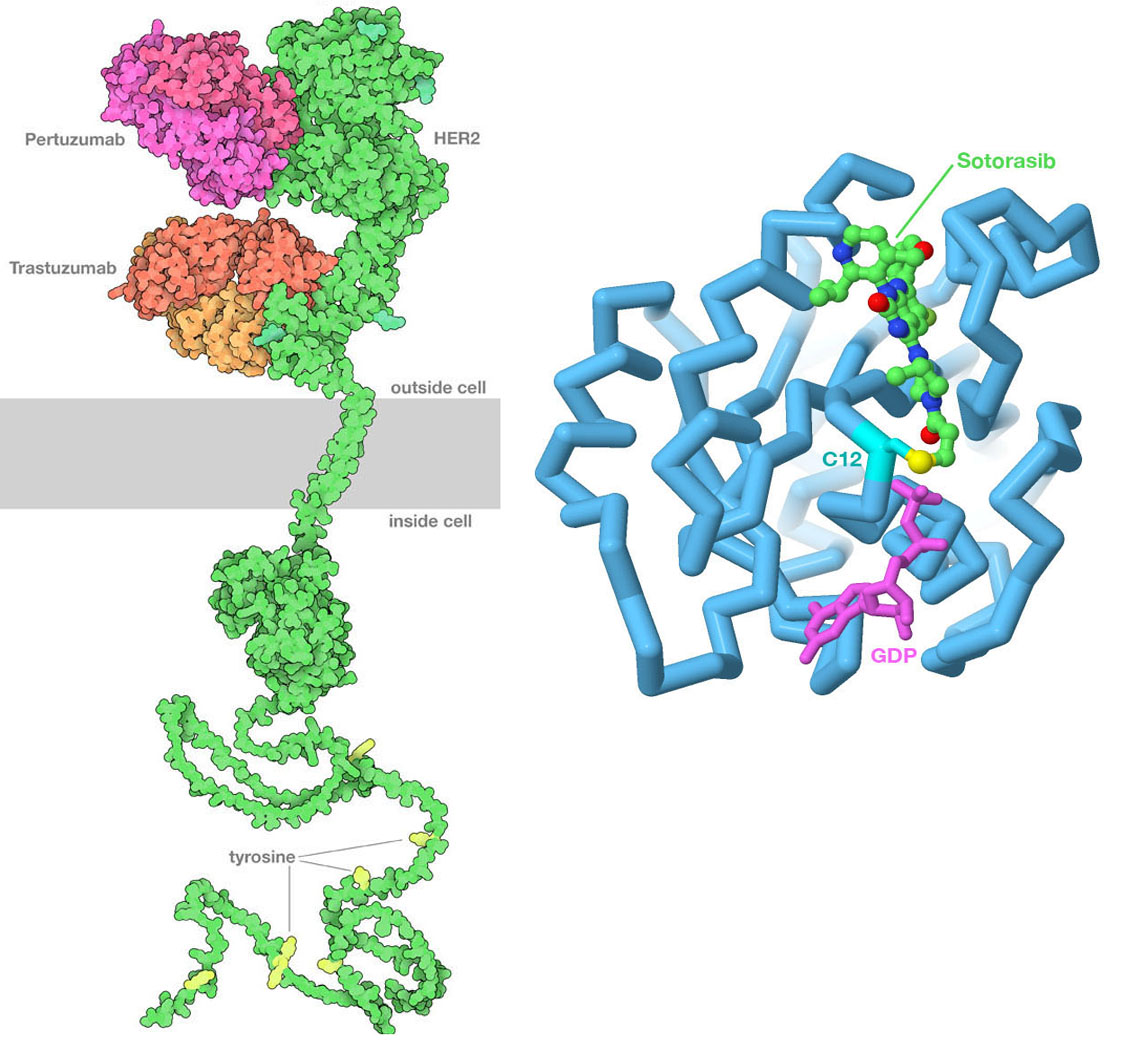
(Left) PDB id 6ogi includes the extracellular domain of HER2 bound to two therapeutic antibodies: pertuzumab and trastuzumab. The antibodies block the formation of active dimers of the receptor, thus blocking the growth signal. The transmembrane domain is from PDB id 2ksi, the kinase domain inside the cell is from PDB ID 3pp0, and the unstructured tail at bottom is from AlphaFold2. (Right) Sotorasib binds covalently to the sulfur atom in cysteine 12 of Ras protein, blocking its action. The drug is shown with carbon atoms in green, the cysteine sulfur is in yellow, and GDP is in magenta. Image created in Jmol using PDB ID 6oim.
Our evolving understanding of cancer has led to the discovery of new approaches to cancer therapy that directly target cancer cells, which is frequently referred to as precision medicine. The key to these approaches is finding genomic or proteomic features that are unique to the cancer cell that can be used to target the therapy to the cancer cell. These features are often proteins on the cell surface that are expressed at much higher levels than in normal cells, making them easy to recognize. The most successful approach so far has been the use of specific antibodies to block key signaling pathways. For example, trastuzumab and pertuzumab are therapeutic antibodies used to treat certain forms of breast cancer that overexpress human epidermal growth factor receptor 2 (HER2), and bevacizumab blocks the signaling molecule VegF involved in formation of new blood vessels.
Ideally, use of precision medicines is contingent on identification of specific changes in the cancer cell genome to ensure that the individual to whom the drug is prescribed is likely to benefit. The G12C variant of the Ras protein described earlier exemplifies this precisely-targeted approach. Sotorasib (sold by Amgen under the brand names Lumakras and Lumykras) is an oral (pill form) anti-cancer medication that is exquisitely selective for G12C KRas. It binds to the enzyme active site by making a covalent bond with the side chain of the aberrant cysteine amino acid residue at position 12. This variant occurs in ~ 14% of non-small cell lung cancers (NSCLCs). Its use was approved by the US Food and Drug Administration (FDA) for the treatment of adult patients with KRas G12C-mutated locally advanced or metastatic NSCLC, as determined by an FDA-approved DNA test.
Researchers are also exploring a wide range of creative ideas for creating targeted therapies that combine two or more molecular functionalities. One of the first ideas was to connect a cancer-binding antibody with a powerful toxin like ricin, to create an antibody-drug conjugate immunotoxin that binds selectively to cancer cells and then kills them. PROTAC molecules are drug-sized molecules that enter cells and link a target protein to the cell’s machinery for protein degradation. CART therapy uses custom cell-surface proteins to engineer an entire T-cell, so that it becomes activated when it binds to cancer cells and destroys them. Innovative medical approaches such as these are the result of structural understanding of the individual molecules, allowing researchers to engineer new chimeric molecules with new functionality.
9. Additional Resources
For more information, see:
-
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646-674.
-
Lambert AW, Pattabiraman DR, Weinberg RA (2016) Emerging biological principles of metastasis. Cell 168, 670-691.
-
Nussinov R, Tsai CJ, Jang H (2020) Ras assemblies and signaling at the membrane. Curr. Op. Struct. Biol. 62, 140-148.
-
Westbrook JD, Soskind R, Hudson BP, Burley SK (2020) Impact of the Protein Data Bank on antineoplastic approvals. Drug Discov. Today 25, 837-850.
Related PDB-101 Learning Resources
All cancer-related PDB-101 resources
Videos about the “Molecular Mechanisms of Cancer” created by high school students
Browse annotations to find PDB structures related to cancer using keywords. Examples:
